Published on 30.07.2025
Bone marrow is a soft tissue located within bones that is responsible for the production of blood cells, including as red blood cells, white blood cells, and platelets. Examples of genetic disorders of the bone marrow include inherited bone marrow failure syndromes. These syndromes are characterized by a total or partial impairment of the production of one or more blood cell types, which can also associate developmental delays and increased susceptibility to develop cancers. The genetic origins and symptoms of these conditions are highly varied, often leading to difficulties or even failure in diagnosis.
In a study published today in The Journal of Clinical Investigation, researchers from the laboratories Human Lymphohematopoiesis and Genome Dynamics and Human Pathologies at the Institut Imagine (Inserm, AP-HP, Université Paris Cité, CIC Biothérapie) report the identification and consequences of an OSM gene mutation in three young patients with bone marrow failure from a consanguineous family.
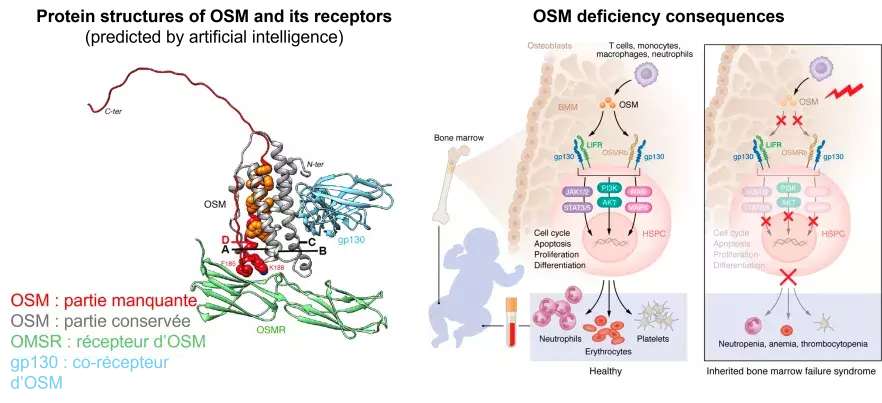
In 2013, patients presenting with severe bone marrow failure characterized by anemia (lack of red blood cells), neutropenia (lack of a type of white blood cell) and thrombocytopenia (lack of platelets), were referred to Patrick Revy's team for suspected telomeropathy. This group of genetic diseases is characterized by the presence of an anomaly in the size or integrity of telomeres (the ends of chromosomes, necessary to protect them from rearrangement or loss of genetic information), a frequent consequence of which is bone marrow failure. In the three patients, telomere analysis revealed no abnormalities, yet whole exome sequencing (a technique for reading the protein-producing parts of genes) identified a mutation in the OSM gene.
Following the identification of the mutation, collaboration between the laboratories and platforms of the Institut Imagine enabled the development of cellular models reproducing the effects of the mutation, thereby facilitating a more profound understanding of its impact. The Institut Imagine's Human Lymphohematopoiesis Laboratory, and more specifically Alexandrine Garrigue, Isabelle André and Chantal Lagresle-Peyrou, made a major contribution to this study. The study was also made possible by the cooperation of patients and their physicians, who facilitated the evaluation of cells harbouring the OSM mutation.
The OSM gene enables the production of the Oncostatin M (OSM) protein, particularly by immune cells (e.g. T lymphocytes, macrophages, etc.). The release of OSM into the body is crucial for its function as a signal, which is achieved by binding to membrane receptors on neighbouring cells. Examples of these receptors include OSMR (OSM receptor) and LIFR (LIF receptor). The binding of OSM to these receptors results in the regulation of various processes, including proliferation and renewal.
Initially, the research teams demonstrated that the patients' immune cells were unable to produce a functional OSM protein. Subsequently, the researchers employed the CRISPR-Cas9 molecular scissors technique to insert the OSM gene mutation identified in the patients into cell models. This confirmed that cells with the OSM mutation are unable to activate their neighboring cells. Finally, by specifically blocking the production of the normally constituted OSM protein in a zebrafish model, Swiss collaborators (Julien Bertrand, University of Geneva) observed symptoms very similar to those of the patients. This finding signifies that OSM deficiency in humans engenders highly specific clinical consequences, albeit severe, which are confined to bone marrow failure. Nevertheless, it will be crucial to monitor the emergence of any additional symptoms over time.
The elucidation of these mechanisms has the potential to unveil novel therapeutic avenues, as the current standard of care for patients with bone marrow failure due to OSM defects frequently involves blood transfusions, which only offer partial symptom relief. The present study proposes the administration of normal OSM proteins as a potential therapeutic intervention to compensate for this deficiency. Finally, this is the very first time that a genetic deficiency of OSM has been described in humans, while many cases of excessive OSM synthesis have already been observed in inflammatory diseases such as multiple sclerosis, Crohn's disease or rheumatoid arthritis. This is why antibodies or inhibitors directed against OSM and OSMR are currently in clinical trials. However, the deleterious consequences of an OSM defect on hematopoiesis described by the Institut Imagine laboratories suggest that the inappropriate use of drugs targeting OSM could have considerable adverse effects.
The article, published in the journal in The Journal of Clinical Investigation, is accompanied by a detailed commentary by two researchers from the Catholic University of Leuven (KU Leuven, Belgium).
References :
Garrigue A, Kermasson L, Susini S, Fert I, Mahony CB, Sadej H, Luce S et al. (2025) Human oncostatin M deficiency underlies an inherited severe bone marrow failure syndrome. J Clin Invest. 135(6):e180981
DOI: 10.1172/JCI180981
Corresponding authors: Patrick Revy & Chantal Lagresle-Peyrou
Delafontaine S & Meyts I (2025) Oncostatin M silence and neopeptide: the value of exploring patients with rare inherited bone marrow failure. J Clin Invest. 135(6):e190955
DOI: 10.1172/JCI190955