Publié le
La moëlle osseuse est un tissu mou situé à l’intérieur des os, qui produit les cellules du sang, comme les globules rouges, les globules blancs et les plaquettes. Des exemples d’atteintes génétiques de la moëlle osseuse sont les syndromes d’insuffisances médullaires héréditaires. Ils sont caractérisés par une atteinte totale ou partielle de la production d’un ou plusieurs types de cellules sanguines, mais peuvent aussi causer des retards de développement et une susceptibilité à développer des cancers. Leurs origines génétiques et leurs symptômes sont très variés, ce qui conduit souvent à des difficultés, voire des absences de diagnostic.
Dans une étude publiée le 17 Mars 2025 dans le journal scientifique JCI, des chercheurs des laboratoires Lympho-hématopoïèse Humaine et Dynamique du Génome et Pathologies Humaines à l'Institut Imagine (Inserm, AP-HP, Université Paris Cité, CIC Biothérapie) rapportent l’identification et les conséquences d’une mutation du gène OSM, chez trois jeunes patientes issues d’une famille consanguine, atteintes d’insuffisance médullaire.
En 2013, les patientes, qui présentaient une défaillance sévère de la moëlle osseuse caractérisée par une anémie (manque de globules rouges), une neutropénie (manque d’un type de globules blancs) et une thrombocytopénie (manque de plaquettes), ont été adressées à l’équipe de Patrick Revy pour une suspicion de téloméropathie. Ce groupe de maladies génétiques est caractérisé par une anomalie de taille ou d’intégrité des télomères (l’extrémités des chromosomes, nécessaires à leur protection contre le réarrangement ou la perte d’informations génétiques), dont une des conséquences fréquentes est l’insuffisance médullaire. Chez les trois patientes, les télomères se sont avérés normaux, mais le séquençage de leur exome (technique permettant de lire les parties de gènes donnant des protéines) a permis d’identifier une mutation du gène OSM.
Après l’identification de cette mutation, la collaboration entre les laboratoires et plateformes de l’Institut Imagine a permis de développer des modèles cellulaires reproduisant les effets de la mutation, pour mieux comprendre son impact sur l’organisme. Le laboratoire Lympho-hématopoïèse Humaine de l’Institut Imagine, et plus spécifiquement Alexandrine Garrigue, Isabelle André et Chantal Lagresle-Peyrou, ont ainsi contribué de façon majeure à cette étude. C’est aussi grâce à la volonté des patientes et à la participation active de leurs médecins que l’évaluation des cellules portant la mutation OSM a pu être complétée.
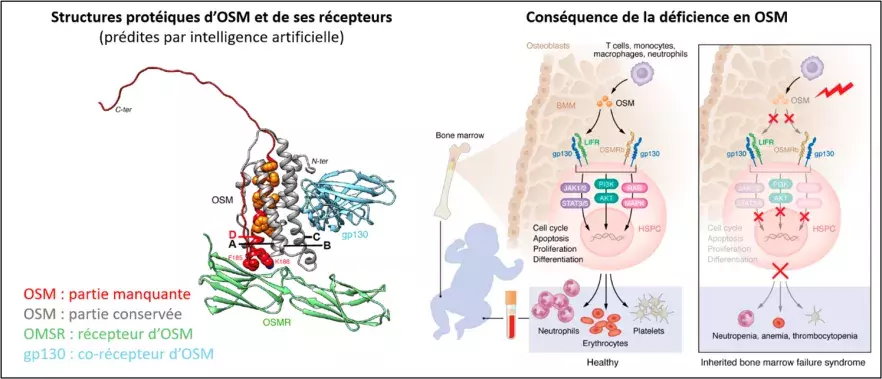
Le gène OSM permet la production de la protéine Oncostatine M (OSM), en particulier par les cellules immunitaires (lymphocytes T, macrophages...). OSM est libérée dans l’organisme afin de servir de signal en se fixant aux récepteurs membranaires de cellules voisines, tels qu’OSMR (récepteur d’OSM) et LIFR (récepteur de LIF), pour, par exemple, réguler leur prolifération et leur renouvellement.
Dans un premier temps, les équipes ont montré que les cellules immunitaires des patientes sont incapables de produire une protéine OSM normale. Puis, grâce à la technique des ciseaux moléculaires CRISPR-Cas9, les chercheurs ont inséré dans des modèles cellulaires la mutation du gène OSM identifiée chez les patientes. Ils confirment ainsi que les cellules portant la mutation OSM sont incapables d’activer leurs cellules voisines. Enfin, en bloquant spécifiquement la production de protéine OSM normalement constituée dans un modèle de poisson zèbre, les collaborateurs suisses (Julien Bertrand, Université de Genève) ont observé des symptômes très similaires à ceux des patientes.
Ils montrent ainsi pour la première fois qu’une déficience en OSM chez l’humain induit des conséquences cliniques très spécifiques, qui, bien que sévères, sont limitées à une insuffisance médullaire. Il sera cependant important de s’assurer que d’autres symptômes n’apparaissent pas au cours de temps.
La compréhension de ces mécanismes ouvre de nouvelles pistes de traitement, car à l’heure actuelle ces patients atteints d’insuffisance médullaire causée par un défaut d’OSM doivent fréquemment subir des transfusions sanguines, qui ne soulagent que partiellement les symptômes. Aujourd’hui, il est possible d’envisager la possibilité d’apporter à ces patients des protéines OSM normales pour combler cette déficience. Enfin, c’est la toute première fois qu’une déficience génétique en OSM est décrite chez l’humain, alors que de nombreux cas de synthèse excessive d’OSM ont déjà été observés dans les maladies inflammatoires telles que la sclérose en plaque, la maladie de Crohn ou encore la polyarthrite rhumatoïde. C’est pourquoi des anticorps ou des inhibiteurs dirigés contre OSM et OSMR sont en cours d’essais cliniques. Cependant les conséquences délétères d’un défaut d’OSM sur l’hématopoïèse décrites par les laboratoires de l’Institut Imagine suggèrent que l’utilisation inadaptée de médicaments ciblant OSM, pourrait avoir des effets indésirables considérables.
L’article, publié dans la revue JCI, est accompagné d’un commentaire détaillé de deux chercheuses de l’université Catholique de Louvain (KU Leuven, Belgique).
Références :
Garrigue A, Kermasson L, Susini S, Fert I, Mahony CB, Sadej H, Luce S et al. (2025) Human oncostatin M deficiency underlies an inherited severe bone marrow failure syndrome. J Clin Invest. 135(6):e180981
DOI: 10.1172/JCI180981
Corresponding authors: Patrick Revy & Chantal Lagresle-Peyrou
Delafontaine S & Meyts I (2025) Oncostatin M silence and neopeptide: the value of exploring patients with rare inherited bone marrow failure. J Clin Invest. 135(6):e190955
DOI: 10.1172/JCI190955