Laurent Abel
Human genetics of infectious diseases : complex predisposition
Published on 22.10.2025
Presentation

Contact
(+33) 1 42 75 43 56
Our group aims to identify the main genes and the corresponding variants involved in the determinism of common infectious diseases. It is also involved in the development of statistical methods in human genetics, since data analyses often raise methodological issues that then we seek to resolve. In particular, we have developed several approaches to improve and optimize the analysis of next generation sequencing (NGS) data. Our studies of infectious diseases mainly focus on infections caused by virulent mycobacteria and certain oncogenic viruses. Our main results over recent years include:
1. In leprosy, after the identification by positional cloning of two major leprosy susceptibility variants (in PARK2/PACRG and LTA genes), we successfully replicated and refined the role of some HLA-C alleles, as well as the role of variants in genes also involved in Crohn’s disease, validating the striking overlap in the genetic control of this disease with leprosy.
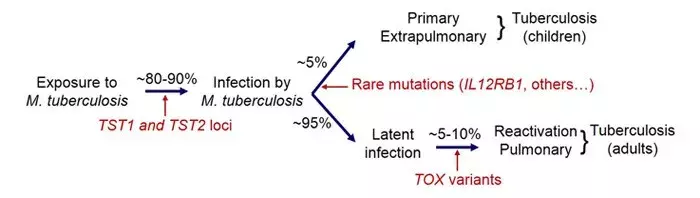
2. In tuberculosis (TB), we identified variants in the gene TOX that are strongly associated with the development of early- onset pulmonary TB (before 25 years of age). Moreover, we discovered additional cases of Mendelian predisposition in disseminated forms of TB in children, due to mutations in TYK2 in particular. Finally, we recently showed replication of the TST1 locus involved in resistance to TB infection in populations of different ethnic origin.
3. In Hepatitis C virus (HCV) infection, we conducted the first genome-wide association study (GWAS) on liver fibrosis caused by chronic HCV infection. We identified several susceptibility loci for HCV-induced liver fibrosis related
to the apoptosis pathway, providing new insights into the mechanisms underlying fibrosis development and paving the way for novel therapeutic strategies.
4. From a methodological point of view, we extended family- based association tests to accommodate variants that are imputed in large GWAS. More recently, we are developing methods to facilitate the interpretation and the identification of disease-causing mutations from NGS data of patients with the same disease. We also showed that whole genome sequencing (WGS) was more powerful than whole exome sequencing (WES) for detection of exonic variants.
We are extending our work on TB and leprosy by studying new phenotypes (subjects highly resistant to TB infection and reversal reactions in leprosy) and collecting new samples.
We have an ongoing project on the genetic basis of Buruli ulcer, the third most common mycobacterial disease. We will continue our work on the most severe HCV- and HBV- related complications such as liver cirrhosis and hepato-carcinoma. Our strategy combines a candidate gene/pathway strategy with genome-wide (GW) approaches. In particular, we are now investigating the role of rare variants with strong individual effects using WES and WGS studies which needs the development of specific analysis methods. All these projects are being performed in large field studies that we are coordinating and involve many collaborators.
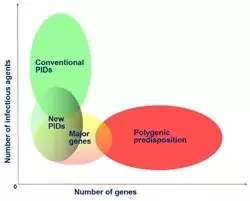
Together with the other research group in the laboratory (JL Casanova), we are also investigating the genetic control of some of these infections from the perspectives of both Mendelian predisposition to rare phenotypes (e.g. severe TB or fulminant hepatitis) and complex predisposition to common phenotypes (e.g. pulmonary TB or HCV/HBV infection). The identification of host genes involved in human infectious diseases will provide new keys to understanding the pathogenesis mechanisms underlying disease development, with potentially major practical implications for the control of infectious diseases.
Team






To read also
-

Research Acceleration
09/03/2023
Identification of a new gene involved in severe allergies and eczema
-

Research Acceleration
01/08/2022
Les causes génétiques des infections graves à staphylocoques
-

Research Acceleration
19/05/2022
Covid-19 and anti IFN1 autoantibodies: the influence of age on mortality
-

Research Acceleration
26/11/2021
Genetics, a compass to explain severe forms of viral infections

Resources & publications
-
 2020Journal (source)Science
2020Journal (source)ScienceInborn errors of type I IFN immunity in patients with life-threatening COVID-19.
-
2020Journal (source)Science
Auto-antibodies against type I IFNs in patients with life-threatening COVID-19.
-
Journal (source)Proc. Natl. Acad. Sci. U.S.A.
Common homozygosity for predicted loss-of-function variants reveals both redu...
-
2020Journal (source)J. Clin. Invest.
Inherited human IFN-γ deficiency underlies mycobacterial disease.
-
2019Journal (source)J. Exp. Med.
Inherited IFNAR1 deficiency in otherwise healthy patients with adverse reacti...
-
2019Journal (source)Front Genet
Identification of an Endoglin Variant Associated With HCV-Related Liver Fibro...
-
Journal (source)Proc. Natl. Acad. Sci. U.S.A.
Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients i...
-
2019Journal (source)Front Immunol
CDG: An Online Server for Detecting Biologically Closest Disease-Causing Gene...
-
2019Journal (source)Hum. Mol. Genet.
A purely quantitative form of partial recessive IFN-γR2 deficiency caused by ...
-
2019Journal (source)Nat. Rev. Immunol.
A novel genetic architecture of infectious diseases.
-
Journal (source)Proc. Natl. Acad. Sci. U.S.A.
Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form ...
-
2019Journal (source)J. Exp. Med.
Severe influenza pneumonitis in children with inherited TLR3 deficiency.
-
2019Journal (source)Bioinformatics
PopViz: a webserver for visualizing minor allele frequencies and damage predi...
-
Journal (source)Proc. Natl. Acad. Sci. U.S.A.
Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form ...
-
2019Journal (source)Sci Immunol
Tuberculosis and impaired IL-23-dependent IFN-γ immunity in humans homozygous...
-
2019Journal (source)PLoS ONE
Prevalence and risk factors for latent tuberculosis infection among healthcar...
-
Journal (source)Proc. Natl. Acad. Sci. U.S.A.
Pleiotropic effects for Parkin and LRRK2 in leprosy type-1 reactions and Park...
-
2019Journal (source)Nucleic Acids Res.
SeqTailor: a user-friendly webserver for the extraction of DNA or protein seq...
-
2019Journal (source)Sci Rep
An eQTL variant of ZXDC is associated with IFN-γ production following Mycobac...
-
2019Journal (source)Elife
IRF4 haploinsufficiency in a family with Whipple's disease.
-
Journal (source)Proc. Natl. Acad. Sci. U.S.A.
Blacklisting variants common in private cohorts but not in public databases o...
-
2019Journal (source)Lancet Infect Dis
Genetics of human susceptibility to active and latent tuberculosis: present k...
-
2018Journal (source)Semin. Immunol.
Human genetics of infectious diseases: Unique insights into immunological red...
-
2018Journal (source)N. Engl. J. Med.
HCV-Associated Liver Fibrosis and HSD17B13.
-
Journal (source)Proc. Natl. Acad. Sci. U.S.A.
Incomplete penetrance for isolated congenital asplenia in humans with mutatio...
-
2018Journal (source)PLoS Negl Trop Dis
Microdeletion on chromosome 8p23.1 in a familial form of severe Buruli ulcer.